全国统一服务热线13433919180

全国统一服务热线13433919180

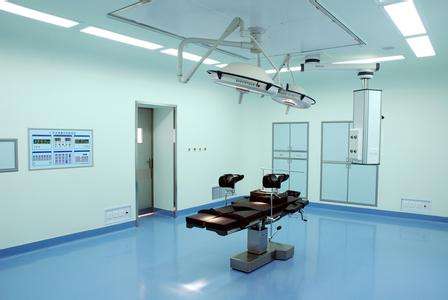
医疗器械GMP洁净车间,医疗器械GMP净化工程,医疗器械GMP洁净工程对非无菌植入性医疗器械或使用前预期灭菌的医疗器械,如果通过确认的产品清洁、包装过程,能将污染降低并保持一致的控制水平,医疗器械GMP车间施工,应建立一个受控的环境来包含该确认的清洁和包装过程。生产企业可参照YY0033-2000标准或自行验证并确定产品的生产洁净级别
3、应对受污染或易于污染的产品进行控制。应对受污染或易于污染的产品、工作台面或人员建立搬运、清洁和除污染的文件。

医疗器械GMP车间工程设计的规范参照:
1、国际标准《ISO/DIS 14644》
2、洁净室厂房设计规范《GB50073-2001》
3、医疗器械包装车间洁净室厂房规范《GMP-97》
4、药品生产质量管理规范《GMP-98》
5、洁净室施工及难收规范《JGJ 71-90》
6、美国联邦标准《FS209E-92》
7、通风与空调工程施工及验收规范《GB 50243-2002》
医疗器械净化工程建设中需考虑以下问题:
1. 医疗器械包装车间洁净室工程所需要的净化材料;
2. 医疗器械厂房洁净室及医疗器械包装车间洁净室工程的设计、安装、调试、维护等综合服务;
3. 医疗器械包装车间洁净室工程空调净化部分:

净化车间温度和相对湿度:
无菌医疗器械在无特殊规定时,通常要求温度在法规标准检测Standard and Testing18~28℃,湿度在 45%~65%,企业一般都可以控制在要求内。如在动态监测中发现达不到要求,可能是室内有产热大的仪器设备。
净化车间风量、换气次数、静压差:
在洁净室体积确定的情况下,换气次数由该室的送风量决定,而静压差取决于房间的送风量与回风量、排风量的差值。系统总送风量、新风量、总排风量和对外压差可以通过调整风机频率转速或总阀门开启度来实现,各房间的风量和压力则可通过调整分支管路阀门开度来实现。
净化车间悬浮粒子、浮游菌、沉降菌:
测试条件如不能满足规定的环境参数 ( 温湿度、风速、换气次数、静压差在规定范围之内 ) 要求,关键项目悬浮粒子、浮游菌或沉降菌的测试结果应视为无效。由于温度、相对湿度、风速、换气次数、静压差共同构成了洁净室的微气候,是洁净室维护正常与否的重要指征,可将关键工序关键项目测试修订为关键工序全性能测试。只有这样,才能全面、系统监测生产洁净室,为确保洁净室性能监测的数据科学性、准确性,测试部门在进行关键项目悬浮粒子、微生物测试时,应同时进行温度、相对湿度、换气次数、静压差等前提条件的测试。
微信二维码
 手机二维码
手机二维码
Copyright©2012-2019 广东坤灵净化设备有限公司 All rights reserved. 备案号:粤ICP备2021061685号 网站统计